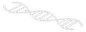
Multiple single-gene biomarkers have already been identified and used in the clinics. However, multiple oncogenes or tumor-suppressor genes are involved during the process of tumorigenesis for each individual patient. Additionally, the efficacy of single-gene biomarkers is limited by the extensively variable expression levels measured by high-throughput assays. Inspired by the success of Gene Set Enrichment Analysis (GSEA), in this study, we advocate for the use of individual-level expression patterns of pre-defined pathways or gene sets as biomarkers. We devised a novel computational method named iPath to identify, at the individual sample level, which pathways or gene sets significantly deviate from their norms.
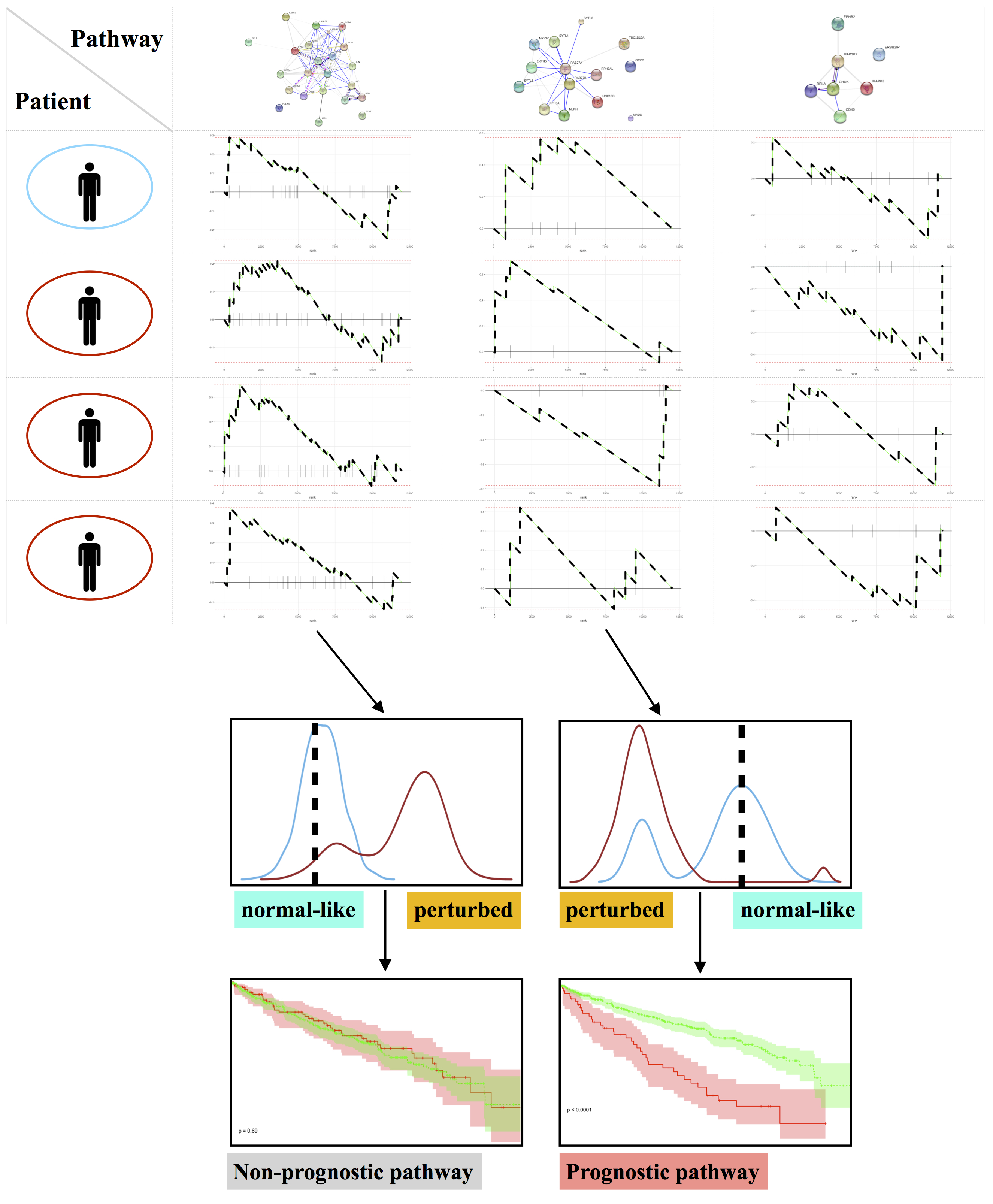
Determining the sample size for adequate power to detect statistical significance is a crucial step at the design stage for high-throughput experiments. We propose POWSC, a simulation-based method, to provide power evaluation and sample size recommendation for single-cell RNA sequencing analysis. POWSC consists of a data simulator that creates realistic expression data, and a power assessor that provides a comprehensive evaluation and visualization of the power and sample size relationship. The data simulator in POWSC outperforms other two state-of-art simulators in capturing key characteristics of real datasets. The power assessor in POWSC provides a variety of power evaluations including stratified and marginal power analyses for two forms of DE under different comparison scenarios.
Power Evaluation and Sample Size Recommendation for Single Cell RNA-seq.
Bioinformatics, June 2020
(code) (talk slides@gmds) (talk recorded@gmds)
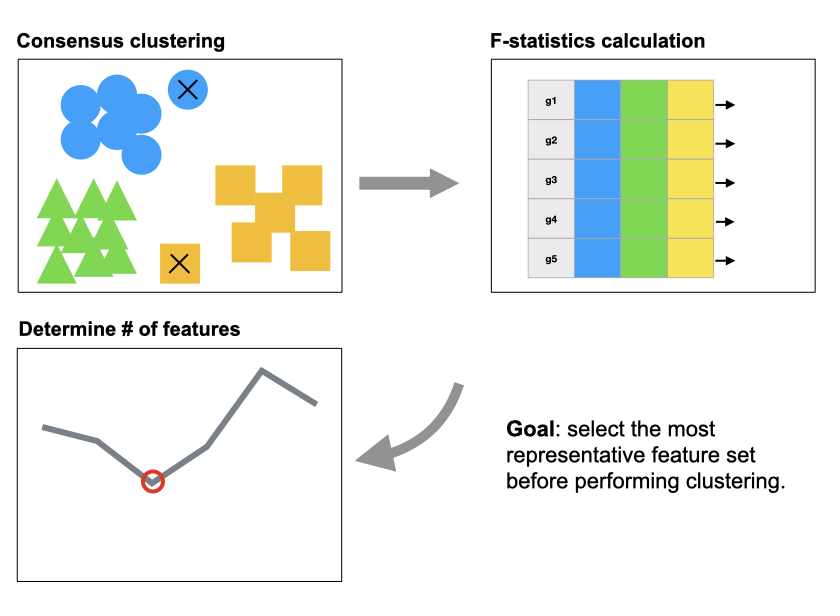
Cell clustering is one of the most important and commonly performed tasks in single-cell RNA sequencing (scRNA-seq) data analysis. An important step in cell clustering is to select a subset of genes (referred to as “features”), whose expression patterns will then be used for downstream clustering. A good set of features should include the ones that distinguish different cell types, and the quality of such set could have significant impact on the clustering accuracy. All existing scRNA-seq clustering tools include a feature selection step relying on some simple unsupervised feature selection methods, mostly based on the statistical moments of gene-wise expression distributions. In this work, we carefully evaluate the impact of feature selection on cell clustering accuracy. In addition, we develop a feature selection algorithm named FEAture SelecTion (FEAST), which provides more representative features. We apply the method on 12 public scRNA-seq datasets, and demonstrate that using features selected by FEAST with existing clustering tools significantly improve the clustering accuracy.
FEAST: a computational algorithm to optimize the most representative feature set before performing clustering.
Briefings in Bioinformatics, Feb 2021
(code)
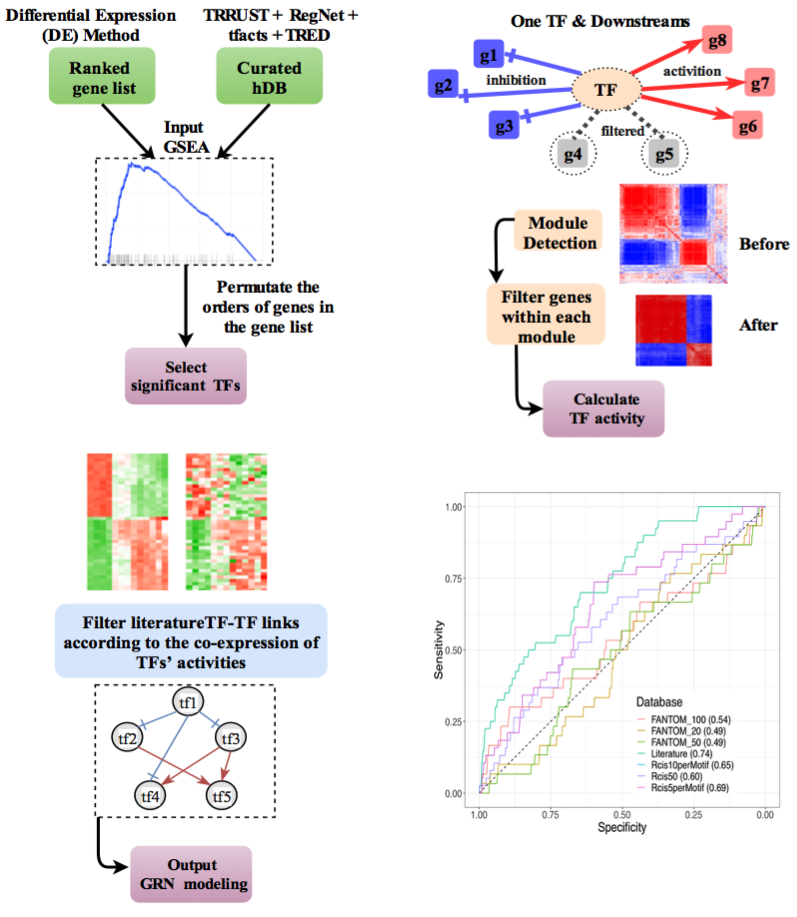
We developed a computational algorithm for constructing transcription-factor-based network, named NetAct, using both transcriptomic data and literature-based transcription factor-target databases. Combining both resources allows us to identify regulatory genes and links specific to the data and fully take advantage of the existing knowledgebase of transcriptional regulation.The algorithm is unique in (1) inferring the activities of regulators for individual samples using the gene expression of their targeted genes, (2) identifying the regulatory interactions between two regulators based on the activity instead of the mRNA expression, (3) integrating mathematical modeling for in silico validation.
NetAct: a computational algorithm to construct core transcription factor regulatory network using gene activity.
Genome Biology, Dec 2022
(code)
Cell Stem Cell,